Authors: Luca Fiore, Arianna Antinucci, Giorgia Leotta, Laura Fabiani, Alessandro Iannini, Pierluca Galloni, Riccardo De Santis, Andrea Ciammaruconi, Giorgia Grilli, Elisa Recchia, Florigio Lista, Fabiana Arduini
A B S T R A C T In the era of sustainability, the use of natural compounds as antimicrobial compounds is the rational selection to avoid the release of pollutants into the environment. Among natural compounds, essential oils are characterized by reliable antimicrobial activity and their use is estimated to grow in the future, thus their detection is an asked point. Herein, we report an electrochemical reagent-free paper-based device for the detection of essential oils, namely thymol, eugenol, and carvacrol by adding 5 µL of solution onto the electrode. We functionalized the working electrode with carbon black by drop casting, demonstrating for the first time the proved sensitivity in essential oil detection using this affordable nanomaterial. To deliver a reagent-free device, the paper-based electrode was loaded with the working buffer for asking the end-user only the addition of the sample. This sensor detected the selected essential oils in a dynamic linear range of up to 16 ppm, with a detection limit equal to 0.1, 0.1, and 0.2 ppm for thymol, eugenol, and carvacrol, respectively. Moreover, the sensor’s sustainability was evaluated using the RGBfast method, highlighting its whiteness compared to conventional chromatographic techniques. The reliable results obtained using the paper-based electrochemical sensor demonstrated the versatility, eco-friendliness, and practicality of this sensing tool, enlarging its use in essential oil detection.
How Simulations Help Understand Their Antimicrobial Potential
Molecular Dynamics (MD) simulations are like a virtual microscope, that allows researchers to observe the movements and interactions of molecules at the atomic level. By applying the fundamental laws of physics, MD simulations predict how atoms and molecules move and interact, which help researchers understand and interpret biological processes, design new drugs, and develop materials with improved and quite often tailored properties.
Antimicrobial peptides have gained significant traction in the past two decades as alternative antibacterial agents through mechanisms involving interactions with both the membrane and intracellular targets, making it harder for antimicrobial resistance to develop, compared to conventional antibiotics. Can you imagine watching a tiny antimicrobial peptide as it moves through water towards a bacterial membrane and attacking it? This is exactly the power of molecular simulations!
As part of the RELIANCE project, the University of Patras team (UPAT) is using MD simulations to investigate the structure and morphology of keratin-derived antimicrobial peptides (KAMPs), namely small proteins originating from humans or chickens with the potential to fight drug-resistant bacteria. The KAMPs under study were first investigated experimentally for their antimicrobial properties by Tam et al.1 and Paul et al.2 In our simulations, we focus particularly on KAMP-10, -10GA, -18C, -18N, -19, -19sc, and -36, as well as on Pw-Antibac123, whose amino acid sequence spans from 1 to 45 residues.1,2 Initially, we explore properties such as their secondary structure and 3D chain conformation in aqueous solutions under dilute and semi-dilute conditions. In the latter case, the aggregation propensity of these short peptides is examined as the result of strong inter-peptide interactions. Next, we explore their interactions with Gram-positive and Gram-negative bacterial models, and more specifically their inner bilayer.
What have our simulations revealed so far?
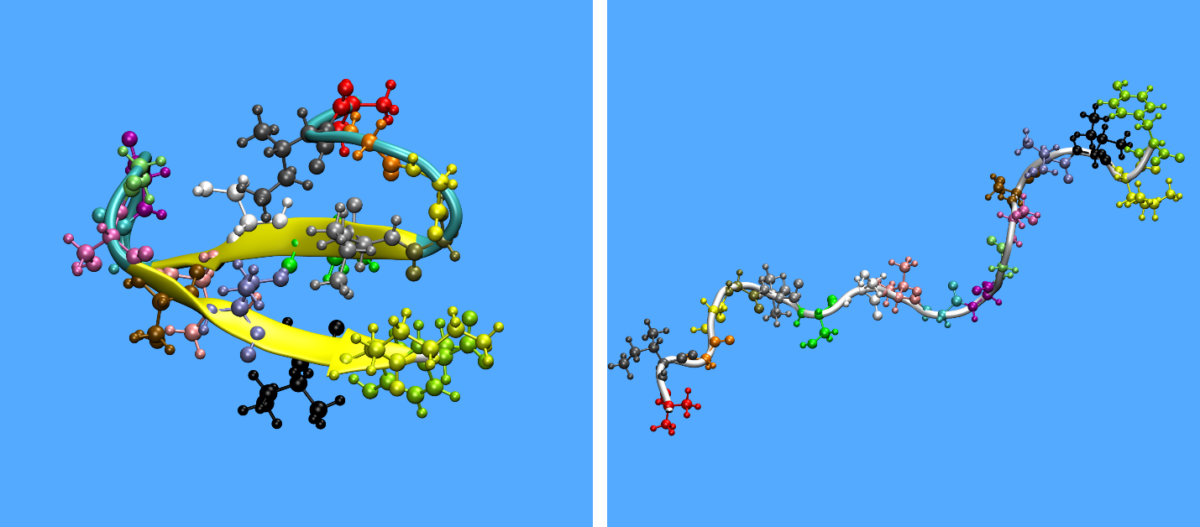
In Figure 1, one can observe the initial and final configuration of a KAMP-18N peptide in dilute conditions.
Figure 1: Initial (left) and final (right) configuration of KAMP-18N in dilute solution. Side chains are coloured by amino acid type while the backbone chain is coloured by secondary structure type.
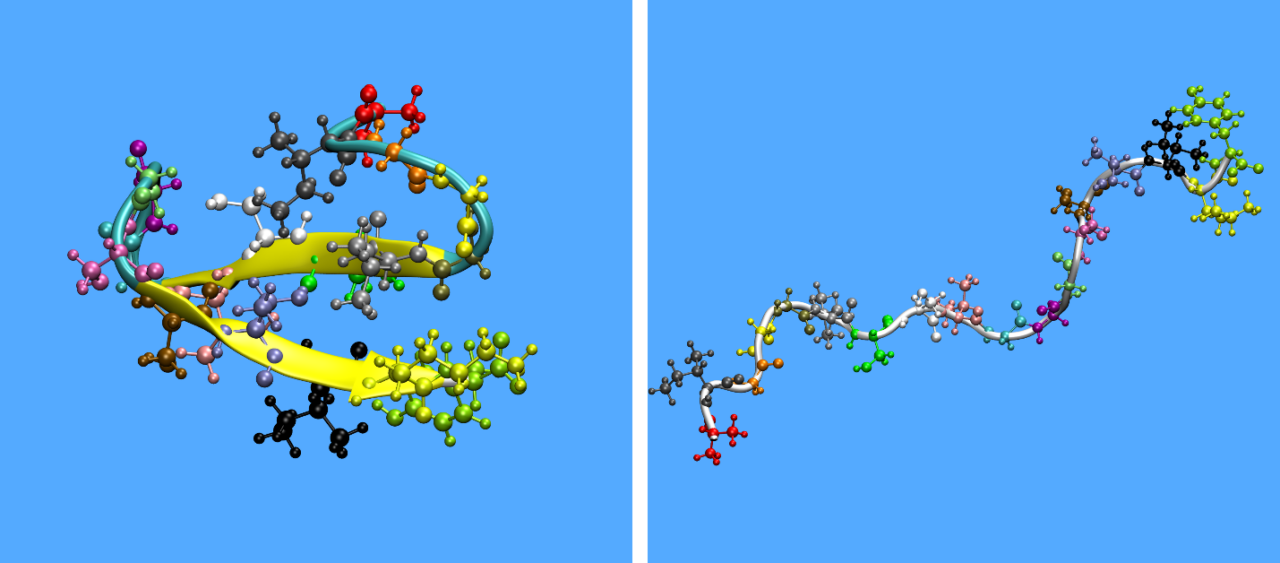
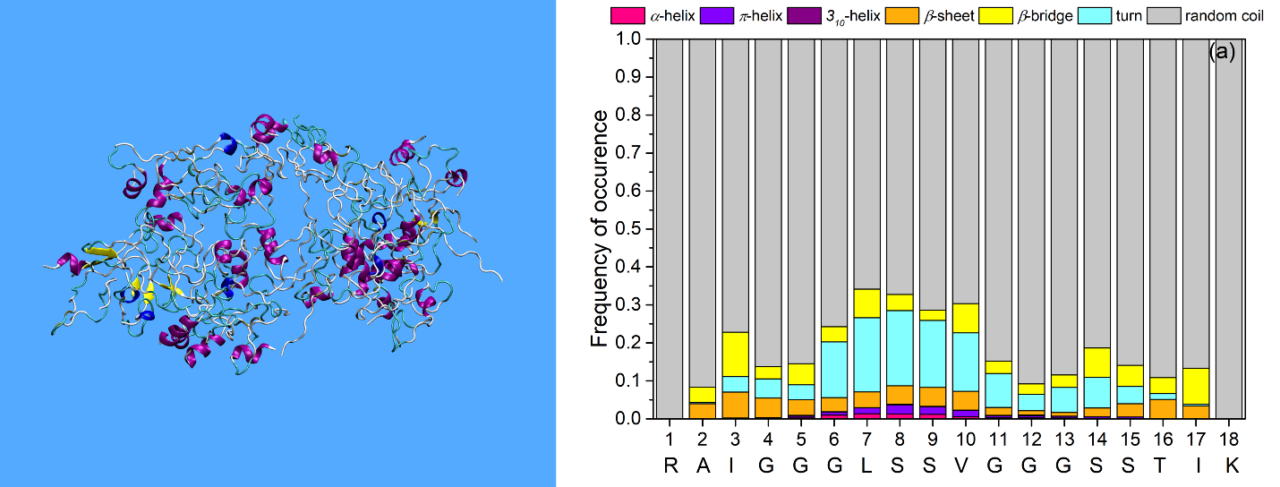
The simulation findings are in very favorable agreement with reported experimental evidence concerning the non-conventional structure of these peptides, further confirming their unique random-coiled configuration in dilute solutions. Turns, helices and β-structures are also observed periodically, but such structural elements are usually unstable, except for Pw-Antivac123 where two stable a-helices are observed. For example, KAMP-19 forms a small α-helix in the intermediate residues 7–9, as also found experimentally3 (Figure 2).
Figure 2: Secondary structure of KAMP-19 as predicted by experiments (Lee et al., 2016) (left) and MD simulations (right).
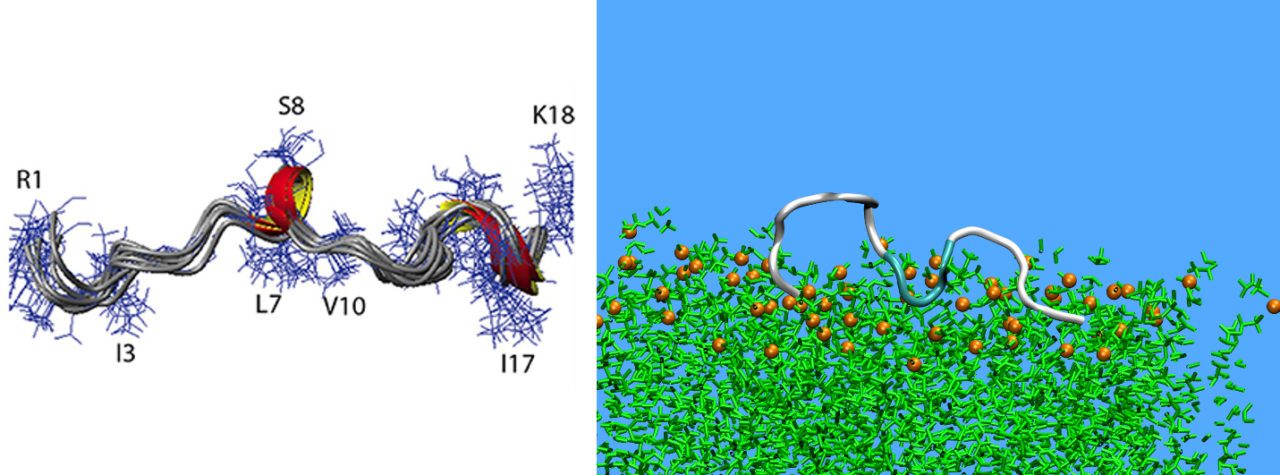
Under semi-dilute conditions, KAMPs from small amorphous aggregates, a process driven by hydrophobic and electrostatic effects. The size and shape of aggregates changes over the simulation time. The formation of a single aggregate is possible (Figure 3), while many peptides may exist as single monomers in the solution. Moreover, the KAMPs maintain again their random coiled structure (Figure 3) as under dilute conditions.
Figure 3: A single aggregate formed by Pw-Antibac123 peptides under semi-dilute conditions (left). Secondary structure distribution of KAMP-18C under semi-dilute conditions (right).
Building on the above findings, the next step is to study the interaction of KAMPs with bacterial membranes. To this, peptides are embedded inside a model lipid bilayer at the beginning of the simulation either in a grid arrangement spanning from one edge to the other or in such a way that a pore is formed at the center of the membrane with an additional grid placed above but close to its upper surface. We observed that, for most of the KAMPs, the pores built in the middle of the membrane are maintained for as long as the simulations are running (e.g., for about one microsecond of time, 1μs) without any peptides observed to escape (Figure 4). For the case where peptides were placed initially inside the lipid bilayer, the peptides also remain embedded in the full course of the simulation, exhibiting a tendency to form small aggregates inside the bilipid.
Figure 4: Initial and final configuration of a KAMP-18N peptides pore inside a Gram-positive membrane with a grid of peptides above the membrane. Peptides are represented as colorful stripes.
What’s next? Future directions of our research
Efforts are currently in progress to learn more about the exact mechanisms of action of KAMPs. With this regard, we are building more realistic systems. For example, by placing above the bilipid membrane a peptidoglycan (PG) layer,4 which is a key component of the bacteria membrane. We are also studying the evolution of systems containing KAMP aggregates embedded in the bacterial membrane to investigate conditions which can cause membrane damage, as the concentration of peptides according to the bibliography is a critical factor that can lead to membrane disruption.
Bibliography
1. Tam, C., Mun, J. J., Evans, D. J. & Fleiszig, S. M. J. Cytokeratins mediate epithelial innate defense through their antimicrobial properties. Journal of Clinical Investigation 122, 3665–3677 (2012).
2. Paul, T. et al. Enzymatic Hydrolyzed Feather Peptide, a Welcoming Drug for Multiple-Antibiotic-Resistant Staphylococcus aureus: Structural Analysis and Characterization. Appl Biochem Biotechnol 175, 3371–3386 (2015).
3. Lee, J. T. Y., Wang, G., Tam, Y. T. & Tam, C. Membrane-active epithelial keratin 6A fragments (KAMPs) are unique human antimicrobial peptides with a non-αβ structure. Front Microbiol 7, (2016).
4. Pokhrel, R., Shakya, R., Baral, P. & Chapagain, P. Molecular Modeling and Simulation of the Peptidoglycan Layer of Gram-Positive Bacteria Staphylococcus aureus. J Chem Inf Model 62, 4955–4962 (2022).
We use cookies to ensure that we give you the best experience on our website. If you continue to use this site we will assume that you are happy with it.